Health Testing
Information for those thinking of breeding their bitches.
BVA eye test and gonioscopy:
DNA tests for PRA and FN: http://www.laboklin.co.uk/laboklin/GeneticDiseases.jsp?catID=DogsGD
AMS: https://www.animaldnadiagnostics.co.uk/
Breeders bible: BOOK OF THE BITCH by J.M.Evans and Kay White available from Amazon.
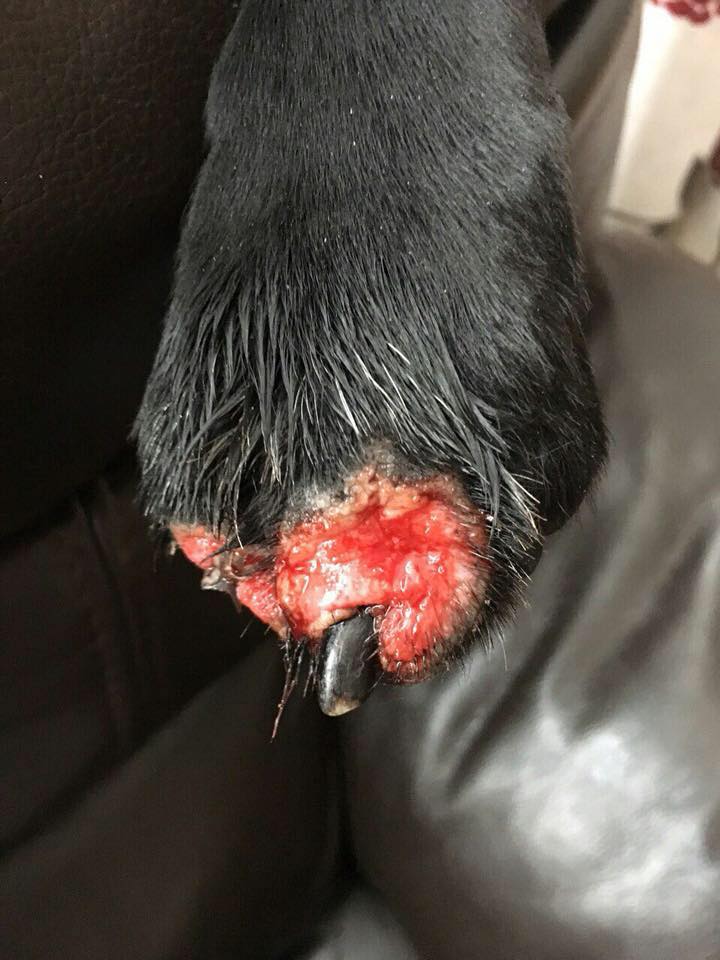
THIS IS AMS
Acral Mutilation Syndrom AMS
Acral mutilation syndrome (AMS) is a rare autosomal-recessive genetic sensory neuropathy of dogs that results in progressive mutilation of the distal extremities[2].
This disease is a variant form of polyneuropathy, similar to ganglioradiculitis, and is characterized by a sensory (afferent) neuropathy in the limbs (stocking-glove sensory loss), and is dissimilar to acral lick dermatitis, which is an obsessive-compulsive disorder of dogs.
AMS results in arrested development of primary sensory neurons followed by progressive postnatal degeneration, with similar neuropathology as is witnessed with Navajo neurohepatopathy[3] in humans.
This disease has been reported in Miniature Schnauzers, German Short-Haired Pointers, English Pointers, English Springer Spaniels and French Spaniels[4].working cocker spaniels.
Clinically affected dogs present with overgrooming and licking of pads and paws to the point of excoriations, ulceration and bleeding. These dogs may be identified soon after birth by their lack of response to acral pinprick or compression. Affected pups are often smaller than unaffected littermates and owners report the pup licking and biting at their paws.
Auto-amputation of claws, digits and footpads occurs in severe cases, with acral changes including swollen reddened paws, paronychia, palmar and plantar ulceration, nail loss and painless fractures.
Neurological examination usually reveals acral analgesia with a variable proximal extent. Tendon reflexes remain intact. Signs of proprioceptive, somatic motor or autonomic impairment have not been observed[1].
In dogs, single or multiple feet can be affected and affected animals can walk on their severely mutilated feet without evidence of pain, lameness, or ataxia[5].
Diagnosis is usually based on nerve biopsies taken under general anesthesia or at postmortem. Reduction in size of the spinal ganglia is observed grossly at necropsy.
A differential diagnosis would include canine distemper virus and other sensory neuropathies such as ganglioradiculitis.
Mild affected dogs can be treated with anxiolytic drugs such as diazepam and elizabethan collars, but many cases rapidly deteriorate, requiring euthanasia due to deteriorating quality of life.
References
Familiar Nephropathy FN
Dogs with FN develop chronic renal failure usually between 6 months and 2 years of age. The first clinical signs observed include excessive water consumption, excessive urine volume, reduced growth rate or weight loss, poor quality hair coat, reduced appetite and vomiting.
FN in English Cocker Spaniels is an autosomal recessively inherited disease and it means that the disease develops only in individuals, which inherited from their parents both mutant alleles (P/P - positive/positive - m mutant homozygote). An individual, which inherits the mutant allele from one parent (result N/P - negative/positive) is heterozygous and a carrier of the disease; this individual transfers this mutant allele to its offsprings. If both parents are heterozygous (carriers of FN), then theoretically, 25 % of the puppies born will be affected.
PRA-prcd disease information
Late form of Progressive Retinal Athrophy, called PRA-prcd (progressive rod-code degeneration), is just one of all retinal defects.
Rods degenerate at first. Affected dogs become night-blind. This is very often the first symptom that dog owners recognize. Dogs usually have poor sense of directions and they crash in things. Pupil is widely open even when direct ray of light hit the eye (dogs have shining eyes in pictures). Later, cones start degenerating. Final disease symptoms are cataracts and total blindness.
PRA-prcd defect arises after normal photoreceptors development. Degree of degeneration differs in parts of retina. Lower retina part is affected sooner and more than upper part (this is not obvious by ophthalmology examination). Disease recognition should be made during dog adolescence. Clinical diagnosis by electroretinogram (ERG) or opthalmoscopy of PRA-prcd can be difficult. ERG identifies affected animals sooner than opthalmoscopy.
PRA-prcd is a hereditary disease. Causal mutation G1298A in ninth canine chromosome (CFA9) PRA-prcd was recognized. This mutation is inherited as an autosomal recessive trait. That means the disease affects dogs with P/P genotype only. The dogs with P/N genotype are considered carriers of the disease (heterozygotes). In offspring of two heterozygous animals following genotype distribution can be expected: 25 % N/N (healthy non-carriers), 25 % P/P (affected), and 50 % N/P (healthy carriers). Because of high risk of producing affected offspring, mating of two N/P animals (carriers) can not be recommended.
Fucosidosis
Fucosidosis is a hereditary disease that occurs when a dog has a mutation in a gene that codes for the enzyme alpha-fucosidase. This enzyme breaks down complex molecules (polysaccharides) so that they can be recycled and/or removed from a cell. The genetic mutation causes a deficiency of alpha-fucosidase; consequently, complex molecules accumulate inside the cell. This abnormal accumulation eventually interferes with the cell’s ability to function.
Most clinical signs of fucosidosis are due to abnormal storage in the cells of the peripheral and central nervous system. Some organs tolerate the accumulation of macromolecules relatively well, but the nervous system appears to be very sensitive. Clinical signs include both behavioral changes and signs of motor dysfunction that start at one to two years of age. Affected dogs exhibit bizarre behavior patterns, may be aggressive or unusually depressed, and appear to forget previously learned behaviors. An affected dog may resist restraint and appear unsteady on its feet. The dog may also appear blind and deaf and may suffer from gastrointestinal disorders such as dysphagia, regurgitation, and diarrhea. Unlike PFK deficiency, this disease progresses rapidly, and death or euthanasia usually occurs within a few weeks from the onset of clinical signs.
Fucosidosis is inherited as an autosomal recessive trait in show and field English Springer Spaniels. A dog that receives a copy of the mutant gene from both parents will show clinical signs. A dog with one copy of the defective gene is a carrier; it will appear healthy but will pass the mutation to its offspring. Researchers at the University of Pennsylvania have developed a test to detect the deletion that occurs in the gene that codes for alpha-fucosidase. This test can detect whether a dog is affected or is a carrier.
Currently, the disease is most prevalent in the conformation lines in the United Kingdom and Australia. However, cases have also been described in field trial dogs. In the United States, clinical cases have been identified, but the number of carriers is still unknown. Fucosidosis may be an emergent disease in the United States, and breeders should consider screening their animals for the presence of carriers. Any English Springer Spaniel that exhibits multifocal neurological signs, especially a dog descended from English families, should be tested for fucosidosis
Phosphofructokinase (PFK) deficiency is an inherited disease that affects both the field trial and show lines of English Springer Spaniels. M-PFK is an enzyme required for the metabolism of glucose into useable energy. Without the PFK enzyme, some cells, such as muscle cells and red blood cells, cannot produce adequate energy for their needs. Therefore, affected dogs display the following intermittent, clinical signs: weakness, lethargy, exercise intolerance, poor performance, muscle cramps, anemia, jaundice, and dark-colored urine. Dark-colored urine, a hallmark of this disorder, usually appears after strenuous exercise or after excessive barking, panting, and heat exposure.
PFK deficiency is caused by a mutation of the gene that codes for the enzyme M-PFK. Because PFK deficiency is an autosomal recessive trait, a dog must have two copies of the mutated gene (one from each parent) to show clinical signs. A dog with one copy may be healthy but will pass the mutation to its offspring. The mutation has so far been documented in over 100 English Springer Spaniels, especially in field trial lines, but the actual prevalence of the mutation is unknown.
Researchers at the University of Pennsylvania have developed a simple, reliable test to detect the mutation that causes PFK deficiency. This test requires a DNA sample obtained from a cheek swab or blood sample.
The following three results are possible:
- Clear – the dog has two normal PFK genes and does not carry the mutation. The dog will not show clinical signs and will not pass the mutation to its offspring. This dog can be used for breeding.
- Carrier – The dog is heterozygous and has one normal and one mutant gene (allele). The dog will not show any clinical signs; therefore, a carrier can be shown and used for field trials without compromising the dog’s health. A carrier will pass the mutant gene to approximately half of its offspring, producing further carriers and continuing the presence of the mutant gene in the general population. Carrier animals should only be bred if they otherwise meet the health, temperament, and quality criteria for breeding; with full disclosure to the owner of the mated dog of the carrier status; with confirmation that the mated dog is not itself a carrier, as such a breeding would produce affected offspring; and with agreement between both parties to the mating that all puppies will be tested prior to placement with all puppies identified as carriers placed with a spay/neuter requirement.
- Affected – the dog has two copies of the mutant gene (allele). The dog will show intermittent clinical signs of disease and will not perform well in field trials. Further, affected dogs will pass the mutation to 100 percent of its offspring. Affected dogs should never be bred.
The cord1-PRA (Cone-rod dystrophy 1) is an inherited disease of the eye that occurs in English Springer Spaniel, Miniature Long-Haired Dachshunds and Smooth-Haired Dachshunds.
The retina is a thin layer of neural cells that lines the back of the eyeball. The vertebrate retina contains photoreceptor cells (rods and cones) that respond to light. The cones mediate high-resolution vision and colour vision. The rods mediate lower-resolution, black-and-white, night vision.
The degeneration of the retina results in loss of vision, often leading to blindness. There is currently no treatment for the disease. In contrast to rod-cone dystrophies, where firstly, rod cells are affected and secondly, degeneration of the cone cells results in complete blindness of the dog, cone-rod dystrophies are characterised by the relatively early loss of cone photoreceptors.
The earliest ophtalmoscopic signs could appear about six month of age but some dogs with the mutation are not diagnosed until much later in life, so owner may never see behavioural changes and never recognise that the dog can pass the mutation onto its offspring.
Since diagnosis of retinal diseases in dogs may prove difficult, the
genetic test on cord1-PRA helps to diagnose a specific form of a disease
and is also a useful tool for breeders to eliminate the mutated gene
from the dog population.
Cord1-PRA is inherited as an autosomal recessive trait. So there are three conditions a dog can be: it can be clear (genotype N/N or homozygous normal) meaning that it does not carry the mutation and will not develop the cord1-form of PRA. Since it also cannot pass the mutation onto its offspring, it can be mated to any other dog.
A dog which has one copy of the gene with the mutation and one copy without the mutation is called a carrier or heterozygous (genotype N/PRA); while it will not be affected by cord1-PRA, it can pass the mutation onto its offspring and should therefore only be mated to clear dogs.
Dogs that develop this form of PRA have two gene copies with the mutation (genotype PRA/PRA or homozygous affected); they will always pass the mutated gene onto their offspring and should also be mated only to clear dogs